")
Mutation KIF5C
Comprendre cette mutation génétique et ses conséquences.
Quand un gène dévie de la normalité, on dit qu’il est muté ou encore qu’il est porteur d’un variant.
Pour reprendre l’explication simplifiée et imagée de notre neuropédiatre : « un variant est une variation d’un gène, c’est-à-dire qu’il y a une erreur de frappe dans un gène sans qu’on puisse affirmer que cette faute de frappe soit responsable d’une maladie. En pratique, nous avons tous beaucoup de variants en nous qui sont des traits de famille. Si on compare les gènes à des recettes de cuisine, on se transmet les recettes de générations en générations dans les familles, et pourtant, si on regarde mot à mot tout n’est pas écrit pareil entre deux recettes d’une même famille, alors que le plat reste très bon dans les deux familles. Une mutation est une erreur de frappe dans un gène qui a clairement prouvé sa responsabilité dans la causalité de la maladie. »
Si la génétique est une recette de cuisine, l’épigénétique est la qualité des ingrédients et des équipements, les mains qui préparent les aliments et l’environnement dans lequel ils sont fabriqués.
L’épigénétique permet le contrôle génétique par des facteurs externes autres que votre ADN. Le mode de vie, l’environnement, les prises en charges thérapeutiques stimulantes et adaptées sont des facteurs essentiels aux progrès des patients atteints de la mutation KIF5C. Plus les thérapies sont précoces, régulières et nombreuses, plus les patients démontrent leurs capacités à progresser.
Une modification irréversible de l'ADN
À ce jour, 45 gènes KIF regroupés en 15 familles ont été identifiés (on estime même qu’il existe deux fois plus de protéines !). Les gènes de la superfamille des kinésines (KIF) codent pour des protéines motrices qui jouent un rôle fondamental dans le fonctionnement, le développement, la survie et la plasticité du cerveau.
Le gène KIF5C, situé sur le chromosome 2, contient l’information qui va permettre de fabriquer des protéines appartenant à la famille des kinésines. Ces protéines jouent un rôle majeur dans le transport de molécules au sein du système nerveux central.
La mutation qui intervient dans ce gène KIF5C entraîne alors une modification de l’information génétique, entraînant la malformation cérébrale et les dysfonctionnements associés à la mutation du gène KIF5C.
Une anomalie génétique (très) rare
Combien sommes-nous ?
Pour Benjamin, le diagnostic a été posé en mai 2017 mais il nous a fallu attendre novembre 2019 avant de pouvoir retrouver, grâce à un groupe privé Facebook, une famille en Norvège et une famille aux USA.
À ce jour, Benjamin est le seul cas identifié en France. Il y a une trentaine de cas dans le monde dont 17 en Europe.
La carte ci-dessous présente la répartition et le nombre de patients dans les différents pays.
Les patients atteints dans le monde
(zoomer/cliquer sur les icônes pour afficher le nombre de cas)
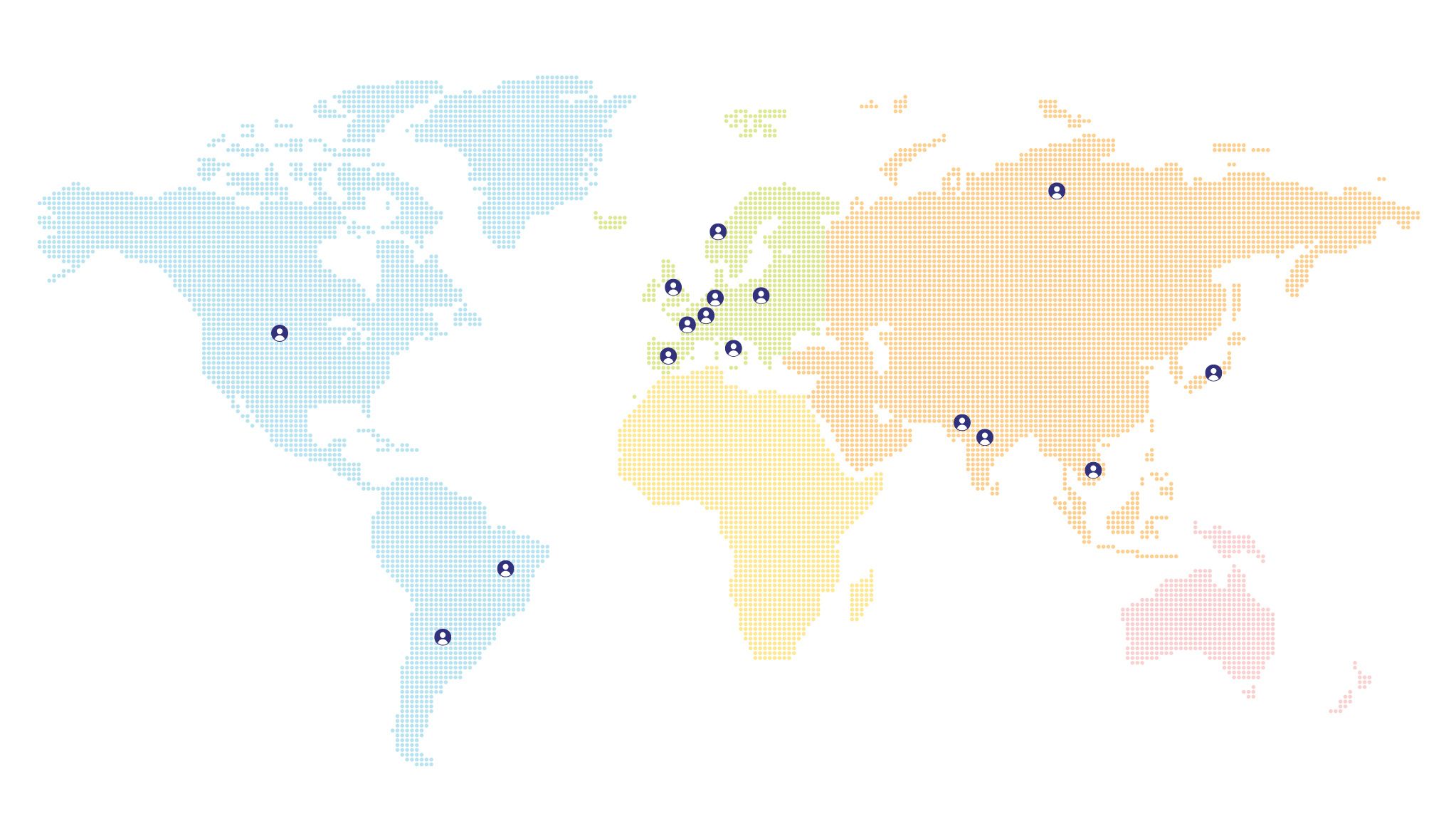
France

1 patient
Allemagne

3 patients
Luxembourg

1 patient
Espagne

1 patient
Italie

2 patients
Norvège

1 patient
Pologne

3 patients
Argentine

1 patient
Brésil

3 patients
Inde

1 patient
Pakistan

1 patient
Russie

1 patient
Japon

1 patient
Vietnam

1 patient
États-Unis

11 patients
Royaume-Uni

6 patients
Mutation du gène KIF5C
Les symptômes
Les enfants atteints de la mutation KIF5C sont pluri ou polyhandicapés. Certains marchent, d’autres pas. La plupart ne parle pas. En grande majorité, ils ont une absence totale d’autonomie et ils doivent être accompagnés dans le quotidien 24h sur 24. Le variant p.Glu237Lys impliquant l’acide aminé Glu237 est le variant le plus représenté à ce jour sur les cas recensés.
Les symptômes identifiés
- Anomalies cérébrales analysées sur les IRM : polymicrogyrie , pachygyrie ainsi qu’une atrophie cérébrale , retard de myéline
- Contact visuel compliqué (regard sur le côté, au plafond)
- Déficience intellectuelle à des degrés variables
- Épilepsie infantile (différents types d’épilepsie incluant habituellement des secousses myocloniques. On constate que ces épilepsies sont souvent réfractaires aux traitements proposés)
- Fatigabilité
- Gestion émotionnelle compliquée avec changements d’humeur soudains
- Hypersensibilité sensorielle (refus de se laver les dents, de se brosser les cheveux, toucher les mains ou les pieds)
- Retard global de développement
- Retard prédominant ou absence de langage (communication non verbale pour la plupart)
- Sourire très présent
- Troubles alimentaires : troubles pondéraux (difficulté dans la prise de poids), gastrostomie, troubles de l’oralité (refus de manger / boire / rejet des textures)
- Troubles des apprentissages
- Troubles du comportement : stéréotypies des mains (applaudissements, mains portées à la bouche), flapping (battement des bras), balancement du corps, tournoiement, automutilation, agressivité, impulsivité, trouble du spectre de l’autisme, trouble déficitaire de l’attention avec ou sans hyperactivité
- Trouble du développement psychomoteur : trouble de la coordination (utilisation limitée au niveau des mains) , instabilité motrice, trouble de la motricité fine, retard ou absence de la marche, hypotonie
- Troubles du sommeil : cycles instables et réveils nocturnes fréquents avec difficulté pour se rendormir
- Troubles gastro-intestinaux : RGO / dysbiose intestinale / constipation chronique
- Troubles ophtalmologiques
Les études scientifiques
Il existe très peu d’études sur la mutation KIF5C. Les chercheurs commencent à lancer des études de cas clinique au vu du nombre de cas recensés qui grandit. Les USA restent le pays avec le plus d’études publiées à ce jour.
Étude menée en 2013
Implication des membres de la famille des kinésines KIF4A et KIF5C dans la déficience intellectuelle et la fonction synaptique.
Introduction
Les gènes de la superfamille des kinésines (KIF) codent pour des protéines motrices qui jouent un rôle fondamental dans le fonctionnement, le développement, la survie et la plasticité du cerveau en régulant le transport de la cargaison le long des microtubules au sein des axones, des dendrites et des synapses. Les études sur les knock-out chez la souris soutiennent ces fonctions importantes du système nerveux. Le rôle des gènes KIF dans la déficience intellectuelle (DI) a jusqu’à présent reçu une attention limitée, bien que des études antérieures aient suggéré que de nombreux gènes ID empiètent sur la fonction synaptique.
Méthode
En appliquant le séquençage de nouvelle génération (NGS) chez les patients atteints de DI, nous avons identifié des mutations pathogènes probables dans KIF4A et KIF5C. Pour confirmer davantage la pathogénicité de ces mutations, nous avons réalisé des études fonctionnelles au niveau de la fonction synaptique dans les neurones primaires de l’hippocampe du rat.
Résultats et conclusions
Quatre mâles d’une même famille avec une mutation perturbatrice dans le KIF4A lié à l’X ont montré une DI légère à modérée et une épilepsie. Une patiente atteinte d’une mutation faux-sens de novo dans KIF5C présentait une DI sévère, une épilepsie, une microcéphalie et une malformation corticale. L’inactivation de KIFA4 dans les neurones primaires de l’hippocampe du rat a modifié l’équilibre entre la transmission synaptique excitatrice et inhibitrice, tandis que la mutation de Kif5c a affecté sa fonction protéique au niveau des synapses excitatrices. Nos résultats suggèrent que les mutations de KIF4A et KIF5C provoquent l’ID en faisant pencher la balance entre l’excitabilité synaptique excitatrice et inhibitrice.
__________
Lien vers l’étude complète : National Library of Medicine
Étude menée en 2017
Les mutations de KIF5C provoquent un trouble neurodéveloppemental de l’épilepsie infantile, une absence de langage et des malformations distinctives du développement cortical.
Introduction
Avec l’avènement des méthodes de séquençage de nouvelle génération (NGS), un nombre croissant de gènes sont identifiés en association avec un large éventail de troubles complexes du développement cérébral (Barkovich et al. 2012). Le diagnostic de malformations corticales sévères, telles que la lissencéphalie classique, est souvent réalisable cliniquement par l’identification d’anomalies caractéristiques de neuroimagerie, ce qui facilite une stratégie de test moléculaire plus ciblée (Guerrini et Dobyns 2014). Cependant, des malformations corticales plus complexes continuent de poser des défis d’un point de vue diagnostique en raison de la variabilité des caractéristiques de neuroimagerie de la malformation cérébrale et/ou de la rareté des types individuels de malformations. Des exemples de telles malformations comprennent la polymicrogyrie (PMG), des types variables de pachygyrie et des formes plus subtiles de dysplasie corticale. Le gène KIF5C a été identifié récemment en 2013 comme une cause rare de malformations corticales caractérisées de manière variable par une pachygyrie régionale ou focale avec ou sans microcéphalie, avec seulement quatre enfants identifiés comme ayant des mutations de ce gène, à ce jour (Jamuar et al. 2014 ; Poirier et al., 2013 ; Willemsen et al., 2014 ; Cavallin et al., 2016). Ici, nous rapportons deux enfants atteints de malformations cérébrales, d’épilepsie infantile et de problèmes neurodéveloppementaux dus à la même mutation de novo KIF5C. En ajoutant nos patients aux quatre individus précédemment rapportés, nous montrons que les mutations de ce gène sont associées à un syndrome neurodéveloppemental distinctif caractérisé par une épilepsie infantile et des malformations cérébrales subtiles mais distinctives reconnaissables par la neuroimagerie.
Matériel et méthode
Les patients ont été inscrits au programme de recherche sur les troubles cérébraux développementaux du Seattle Children’s Research Institute, avec l’approbation de l’IRB. Les chercheurs ont examiné toutes les données cliniques, de neuroimagerie et moléculaires. L’analyse moléculaire a été effectuée à l’aide de panels commerciaux ciblés de nouvelle génération cliniquement disponibles composés de > 1000 gènes associés à l’épilepsie, à la déficience intellectuelle et à l’autisme à l’aide du kit Exome d’Agilent Clinical Research. Le séquençage a été effectué à l’aide du système de séquençage Illumina avec des lectures d’extrémités appariées de 2×100 pb.
Résultats
La neuroimagerie et les caractéristiques cliniques de nos deux patients, ainsi que des individus porteurs de la mutation KIF5C précédemment publiés, sont résumés dans le tableau 1.
__________
Lien vers l’étude complète : National Library of Medicine
Étude menée en 2024
Élargissement du phénotype moléculaire et clinique des patients présentant des variants de novo dans KIF5C : une série de cas de six patients.
Introduction
Des variantes hétérozygotes de novo de la perte de fonction dans le domaine moteur de KIF5C sont associées à un trouble neurodéveloppemental caractérisé par une épilepsie infantile, une dysplasie corticale frontale et des retards de développement, y compris des troubles moteurs et de la parole. Auparavant, seuls trois variants faux-sens de KIF5C étaient connus pour être pathogènes. Nous avons identifié six autres patients présentant des retards de développement significatifs avec des variants hétérozygotes de novo du gène KIF5C (Glu237Val, Thr93Ile, Thr93Asn, Ser90del, Lys92Arg et Glu237Lys), dont quatre variants n’avaient pas été signalés auparavant. Une évaluation fonctionnelle a été réalisée sur des variants de KIF5C marqués par fluorescence exprimés dans des neurones isolés de l’hippocampe. Les variants pathogènes de novo ont montré une fonction motrice significativement réduite par rapport au KIF5C de type sauvage. Nous concluons que les variants pathogènes de novo présentés ont une activité motrice réduite et qu’il est probable qu’il s’agisse de l’étiologie des symptômes des patients compte tenu de la contrainte du gène dans la population. En ajoutant ces patients aux sept patients précédemment rapportés, nous sommes en mesure d’élargir le spectre phénotypique associé aux variants pathogènes de KIF5C. L’évaluation du phénotype neurodéveloppemental d’autres individus présentant des variantes de perte de fonction dans KIF5C est indiquée pour caractériser davantage le spectre des phénotypes associés.
La superfamille de protéines kinésines (KIF) agit comme des moteurs moléculaires dépendants de l’ATP pour faciliter le transport intracellulaire le long des microtubules (Hirokawa et al. 2009), un processus essentiel au développement, au fonctionnement, à la plasticité et à la survie du cerveau (Hirokawa, Niwa et Tanaka 2010). Les protéines KIF régulent le transport de cargaisons telles que les vésicules, les organites, les complexes protéiques, les ARNm et les chromosomes le long des microtubules à l’intérieur des axones, des dendrites et des synapses (Willemsen et al. 2014). Les protéines KIF sont régulées par phosphorylation. Par exemple, c-Jun NH2-la kinase terminale 3 (JNK3) phosphoryle le domaine moteur de KIF5C (l’une des trois variantes également connues sous le nom de kinésine-1 (Miki et al. 2001)), empêchant KIF5C de se lier aux microtubules (Morfini et al. 2009). La perturbation du processus normal de phosphorylation des protéines KIF5C a été suspectée comme un mécanisme physiopathologique pour les maladies neurodégénératives. La répétition anormalement longue de la polyglutamine dans la forme pathogène de la huntingtine augmente l’activité de JNK3, provoquant une hyperphosphorylation de KIF5C, ce qui suggère que la pathologie de la maladie de Huntington pourrait en partie résulter de la diminution du transport de cargaison le long des microtubules par les protéines KIF5 (Morfini et al., 2009).
De même, les variants génétiques qui perturbent directement le fonctionnement du domaine moteur produisent également des phénotypes neurologiques (Duquesne et al. 2020 ; Naim et al., 2022 ; Banerjee et al., 2024 ; Becker et al., 2024). Il a été établi que le gène KIF5C est la cause génétique d’un trouble neurodéveloppemental caractérisé par une épilepsie infantile, une dysplasie corticale frontale et des retards de développement, y compris des troubles moteurs et de la parole, avec sept patients signalés dans la littérature à ce jour (Willemsen et al., 2014 ; Duquesne et al., 2020 ; Jamuar et al., 2014 ; Poirier et al., 2013 ; Cavallin et al., 2016 ; Michels et al., 2017). Le trouble neurodéveloppemental est le résultat de protéines kinésines codées par KIF5C hébergeant un domaine moteur non fonctionnel et perdant la capacité de se lier à l’ATP hydrolysé (Padzik et al. 2016), ce qui altère ou inhibe le transport de la cargaison médié par KIF5C le long des microtubules. Il est intéressant de noter que cinq des sept patients signalés ont présenté des variants impliquant l’acide aminé Glu237, quatre de ces individus partageant le même variant faux-sens, p.Glu237Lys, suggérant un point chaud pour les variants et les corrélations potentielles génotype-phénotype (Michels et al. 2017).
À ce jour, seuls trois variants faux-sens de KIF5C ont été signalés (Willemsen et al. 2014 ; Jamuar et al., 2014 ; Poirier et al., 2013 ; Cavallin et al., 2016 ; Michels et al., 2017). Le gène KIF5C a d’abord été identifié comme étant la cause de la malformation du développement cortical (Poirier et al. 2013). Les personnes touchées présentent généralement un retard neurodéveloppemental, une épilepsie infantile, une déficience intellectuelle et un retard psychomoteur et des problèmes de comportement (Duquesne et al. 2020 ; Banerjee et al., 2024 ; Michels et al., 2017). La malformation du développement cortical peut inclure une polymicrogyrie et une pachygyrie ainsi qu’une atrophie cérébrale (Duquesne et al. 2020 ; Banerjee et al., 2024 ; Michels et al., 2017). Cependant, des signes cliniques tels que l’épilepsie, les retards de développement et les déficiences intellectuelles sont des présentations cliniques courantes pour une grande variété de conditions génétiques et multifactorielles et ne suggèrent donc pas immédiatement une étiologie sous-jacente de KIF5C. L’utilisation croissante des technologies de séquençage de nouvelle génération (NGS) dans le cadre de l’évaluation diagnostique de ces enfants conduit à la découverte de variantes de signification incertaine dans KIF5C. Nous rapportons ici six autres enfants avec des variants de novo dans KIF5C, dont quatre n’ont jamais été signalés auparavant. En ajoutant ces patients aux sept patients précédemment rapportés, le spectre phénotypique associé aux variants pathogènes de KIF5C est élargi.
Méthode
Cette série de cas a été préparée sur la base du protocole #19-0751 du Colorado Multiple Institutional Review Board (COMIRB).
Résultats et conclusions
Quatre mâles d’une même famille avec une mutation perturbatrice dans le KIF4A lié à l’X ont montré une DI légère à modérée et une épilepsie. Une patiente atteinte d’une mutation faux-sens de novo dans KIF5C présentait une DI sévère, une épilepsie, une microcéphalie et une malformation corticale. L’inactivation de KIFA4 dans les neurones primaires de l’hippocampe du rat a modifié l’équilibre entre la transmission synaptique excitatrice et inhibitrice, tandis que la mutation de Kif5c a affecté sa fonction protéique au niveau des synapses excitatrices. Nos résultats suggèrent que les mutations de KIF4A et KIF5C provoquent l’ID en faisant pencher la balance entre l’excitabilité synaptique excitatrice et inhibitrice.
__________
Lien vers l’étude complète : Wiley Online Library
Analyse sur un panel de 30 patients
Les données ci-après sont fournies à titre indicatif par 30 familles. Dans tous les cas, les parents restent les experts de leur enfant et il est montré que les patients atteints de la mutation KIF5C progressent tous quand ils sont stimulés et accompagnés dans des thérapies adaptées. Ces chiffres sont donc pris à un instant T, ils portent sur 30 enfants de 16 pays différents (16 filles âgées de 2 ans à 27 ans et 14 garçons âgés de 16 mois à 19 ans).
Tranches d'âges des patients :
- Enfants de 0 à 3 ans 17%
- Enfants de 4 à 11 ans 63%
- Adolescents de 12 à 18 ans 20%
- Adultes de 19 à 25 ans 10%
- Adultes de plus de 25 ans 7%
%
Ne marchent pas
- Filles 23%
- Garçons 77%
%
Ne parlent pas
- Filles 52%
- Garçons 48%
%
Ne mangent pas solide
- Filles 40%
- Garçons 60%
%
Sont épileptiques
- Filles 50%
- Garçons 50%
Les prises en charge
Comment se faire accompagner ?
Il n’existe aucun traitement spécifique pour cette mutation KIF5C à ce jour. Les parcours des patients sont très différents selon le profil et les besoins des patients, les ressources des familles, le pays, mais on constate que les progrès sont possibles quand les patients sont stimulés et les prises en charge adaptées. Les familles font un travail formidable dans l’accompagnement de la maladie et remplacent souvent les soins manquants. Des accompagnements pluridisciplinaires doivent donc être mis en place le plus tôt possible.
Les accompagnements pris en charge et non pris en charge par la Sécurité Sociale en France :
Pris en charge
- Neuropédiatre / pédiatre
- Endocrinologue
- Généticien
- Ophtalmologue / orthoptiste
- ORL
- Psychiatre
- Kinésithérapeute
- Orthophoniste
- Neurologue
- Médecin spécialisé dans les troubles du sommeil
- Médecin spécialisé dans les troubles alimentaires
- Médecin spécialisé dans les troubles neurodéveloppementaux (TSA, TDAH, TED)
- Infirmière / auxiliaire de vie
Non pris en charge
- Neuropsychologue
- Psychomotricien
- Ergothérapeute
- Psychologue
- Éducateur spécialisé
- Nutritionniste
- Ostéopathe
- Équithérapie / hippothérapie
- Aquathérapie
HAPPY BENJI : en avant, marche ! est une association caritative fondée en 2025 par les parents de Benjamin.
Elle œuvre pour faire reconnaître la maladie due à une mutation du gène KIF5C, fédérer les familles de patients atteints et convaincre le corps médical d’engager des recherches de thérapie génique.
