")
KIF5C mutation
Understand this genetic mutation and its consequences.
When a gene deviates from normality, it is said that it is mutated or that it is carrying a variant.
To repeat the simplified and imaginative explanation of our neuropaediatrician: « a variant is a variation of a gene, i.e. there is a typing error in a gene without being able to say that this typing error is responsible for a disease. In practice, we all have many variations within us that are family traits. If we compare genes to cooking recipes, the recipies are passed down from generation to generation, and yet, if you look word by word, everything is not written the same between two recipes of the same family, while the dish remains very good in both families. A mutation is a typing error in a gene that has clearly proven its responsibility for the causality of the disease. »
If genetics is a cooking recipe, the epigenetics is the quality of the ingredients and equipment, the hands that prepare the food and the environment in which they are made.
Epigenetics allows genetic control by external factors other than your DNA. Lifestyle, environment, stimulating and appropriate therapeutic care are essential factors for the progress of patients with KIF5C mutation. The more early, regular and numerous therapies, the more patients demonstrate their ability to advance.
An irreversible change in DNA
To date, 45 KIF genes grouped into 15 families have been identified (it is even estimated that there are twice as many proteins!). Kinesine superfamily (KIF) genes code for motor proteins that play a fundamental role in the functioning, development, survival and plasticity of the brain.
The KIF5C gene, located on chromosome 2, contains information that will make protein from the kinesine family. These proteins play a major role in the transport of molecules within the central nervous system.
The mutation in this KIF5C gene results in a change in genetic information, resulting in brain malformation and dysfunction associated with the KIF5C gene mutation.
A (very) rare genetic anomaly
How many are we ?
As far as Benjamin is concerned, the diagnosis was made in May 2017 but we had to wait until November 2019 before we could find 2 families with KIF5C mutation abroad.
To date, Benjamin is the only case identified in France. There are about 30 cases in the world, including 17 in Europe.
The map below shows the distribution and number of patients in different countries.
Patients in the world
(zoomer/click icons to display number of cases)
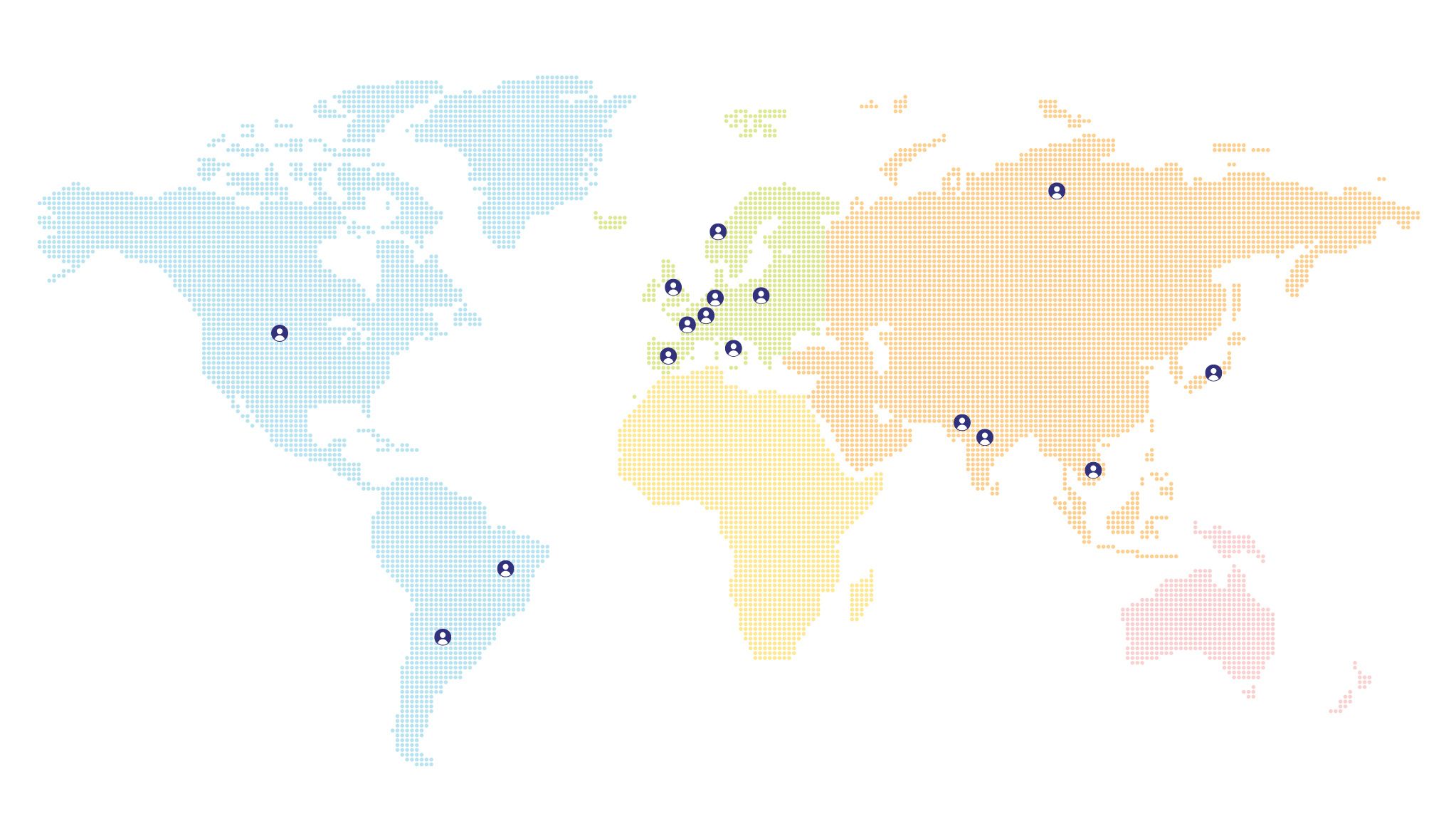
France

1 patient
Germany

3 patients
Luxembourg

1 patient
Spain

1 patient
Italy

2 patients
Norway

1 patient
Poland

3 patients
Argentina

1 patient
Brazil

3 patients
India

1 patient
Pakistan

1 patient
Russia

1 patient
Japan

1 patient
Vietnam

1 patient
United States

11 patients
United Kingdom

6 patients
KIF5C gene mutation
Symptoms
Children with KIF5C mutations are multiple or poly-disabled. Some walk, others do not walk. Most patients do not talk. In the vast majority, they have a total lack of autonomy and must be accompanied in daily life 24 hours a day. The variant p.Glu237Lys involving amino acid Glu237 is the most represented variant to date on the reported cases.
Symptoms identified
- Brain abnormalities analyzed on MRI: polymicrogyry, pachygyria as well as cerebral atrophy, myelin delay
- Complicated visual contact (looking at side, ceiling)
- Intellectual disability to varying degrees
- Infant epilepsy (different types of epilepsy usually including myoclonic tremors. It is found that these epilepsies are often refractory to the proposed treatments)
- Fatigability
- Complex emotional management with sudden mood changes
- Sensory hypersensitivity (refused to wash teeth, brush hair, touch hands or feet)
- Overall development delay
- Predominant delay or absence of language (mostly non-verbal)
- Very present smile
- Eating disorders : weight disorders (difficulty in weight gain), gastrostomy, orality disorders (refusal to eat / drink / rejection of textures)
- Learning disorders
- Behavioural disorders : hand stereotyping (applause, hands carried to mouth), flapping (arm swing), body swinging, spinning, self-mutilation, aggressiveness, impulsivity, autism spectrum disorder, attention deficit disorder with or without hyperactivity
- Psychomotor development disorder : coordination disorder (limited use in the hands), motor instability, fine motor disorder, delay or absence of walking, hypotonia
- Sleep disorders: unstable cycles and frequent night wake-ups with difficulty in returning to sleep
- Gastrointestinal disorders : GERD / intestinal dysbiosis / chronic constipation
- Ophthalmological disorders
Scientific studies
There are very few studies of KIF5C mutation. Researchers are starting to launch clinical case studies given the growing number of reported cases. The USA remains the country with the most published studies to date.
Study conducted in 2013
Involvement of kinesine family members KIF4A and KIF5C in intellectual disability and synaptic function.
Introduction
Kinesine superfamily genes (KIF) code for motor proteins that play a fundamental role in the functioning, development, survival and plasticity of the brain by regulating the transport of cargo along microtubules within axons, dendrites and synapses. Knock-out studies in mice support these important nervous system functions. The role of KIF genes in intellectual disability (ID) has so far received limited attention, although previous studies have suggested that many ID genes interfere with synaptic function.
Method
Applying new generation sequencing (NGS) in ID patients, we identified probable pathogenic mutations in KIF4A and KIF5C. To further confirm the pathogenicity of these mutations, we conducted functional studies of synaptic function in primary rat hippocampal neurons.
Results and conclusions
Four males from the same family with a disruptive mutation in the KIF4A bound to the X showed mild to moderate DI and epilepsy. A patient with novo false-sense mutation in KIF5C had severe ID, epilepsy, microcephaly, and cortical malformation. KIFA4 inactivation in primary rat hippocampus neurons altered the balance between excitatory and inhibitory synaptic transmission, while Kif5c mutation affected its protein function in excitatory synapses. Our results suggest that KIF4A and KIF5C mutations cause ID by tilting the balance between excitatory and inhibitory synaptic excitability.
__________
Link to the complete study : National Library of Medicine
Study conducted in 2017
KIF5C mutations cause a neurodevelopmental disorder of childhood epilepsy, a lack of language and distinctive malformations of cortical development.
Introduction
With the advent of new generation sequencing methods (NGS), an increasing number of genes are identified in association with a wide range of complex brain development disorders (Barkovich et al. 2012). The diagnosis of severe cortical malformations, such as classical smoothencephaly, is often clinically feasible by identifying abnormalities characteristic of neuroimaging, which facilitates a more targeted molecular test strategy (Guerrini and Dobyns 2014). However, more complex cortical malformations continue to pose diagnostic challenges due to the variability of neuroimaging characteristics of brain malformation and/or the scarcity of individual types of malformations. Examples of such malformations include polymicrogyria (PMG), variable types of pachygyria, and more subtle forms of cortical dysplasia. The KIF5C gene was recently identified in 2013 as a rare cause of cortical malformations variablely characterized by regional or focal pachygyria with or without microcephaly, with only four children identified as having mutations in this gene, to date (Jamuar et al. 2014 ; Poirier et al., 2013 ; Willemsen et al., 2014 ; Cavallin et al. 2016). Here we report two children with cerebral malformations, childhood epilepsy and neurodevelopmental problems due to the same novo KIF5C mutation. By adding our patients to the four previously reported individuals, we show that mutations in
Equipment and methodology
Patients were enrolled in the Seattle Children's Research Institute's developmental brain disorder research program, with approval from the BRI. The researchers examined all clinical, neuroimaging and molecular data. Molecular analysis was performed using new-generation, clinically available, targeted commercial panels composed of > 1000 genes associated with epilepsy, intellectual impairment and autism using the Exome d'Agilent Clinical Research kit. Sequencing was performed using the Illumina sequencing system with 2×100 bp paired end readings.
Results
The neuroimaging and clinical characteristics of our two patients, as well as individuals carrying the previously published KIF5C mutation, are summarized in Table 1.
__________
Link to the complete study : National Library of Medicine
Study conducted in 2024
Expansion of the molecular and clinical phenotype of patients with novo variants in KIF5C : a series of six patients.
Introduction
Heterozygous de novo variants of loss of function in the motor domain of KIF5C are associated with a neurodevelopmental disorder characterized by infantile epilepsy, frontal cortical dysplasia and developmental delays, including motor and speech disorders. Previously, only three false-sense variants of KIF5C were known to be pathogenic. We identified six other patients with significant developmental delays with de novo heterozygous variants of the KIF5C gene (Glu237Val, Thr93Ile, Thr93Asn, Ser90del, Lys92Arg and Glu237Lys), four of which had not been previously reported. Functional evaluation was performed on fluorescence-labelled KIF5C variants expressed in isolated neurons of the hippocampus. Novo pathogenic variants showed significantly reduced motor function compared to wild type KIF5C. We conclude that the presented de novo pathogens have reduced motor activity and that it is likely to be the etiology of patient symptoms given the strain of the gene in the population. By adding these patients to the seven previously reported patients, we are able to broaden the phenotypic spectrum associated with the pathogenic variants of KIF5C. The evaluation of the neurodevelopmental phenotype of other individuals with variants of loss of function in KIF5C is indicated to further characterize the spectrum of associated phenotypes.
The kinesine protein superfamily (KIF) acts as ATP-dependent molecular motors to facilitate intracellular transport along microtubules (Hirokawa et al. 2009), a process essential to brain development, functioning, plasticity and survival (Hirokawa, Niwa and Tanaka 2010). KIF proteins regulate the transport of cargoes such as vesicles, organelles, protein complexes, mRNAs, and chromosomes along microtubules within axons, dendrites, and synapses (Willemsen et al. 2014). KIF proteins are regulated by phosphorylation. For example, c-Jun NH2-terminal kinase 3 (JNK3) phosphoryles the engine domain of KIF5C (one of the three variants also known as kinesine-1 (Miki et al. 2001)), preventing KIF5C from binding to microtubules (Morfini et al. 2009). Disruption of the normal phosphorylation process of KIF5C proteins was suspected as a physiopathological mechanism for neurodegenerative diseases. Abnormally long repeats of polyglutamine in the pathogenic form of huntingtin increased the activity of JNK3, causing hyperphosphorylation of KIF5C, suggesting that the pathology of Huntington's disease may in part result from reduced cargo transport along microtubules by KIF5 proteins (Morfini et al. 2009).
Similarly, genetic variants that directly disrupt the functioning of the motor domain also produce neurological phenotypes (Duquesne et al. 2020; Naim et al., 2022; Banerjee et al., 2024; Becker et al., 2024). It has been established that the KIF5C gene is the genetic cause of a neurodevelopmental disorder characterized by childhood epilepsy, frontal cortical dysplasia and developmental delays, including motor and speech disorders, with seven patients reported in the literature to date (Willemsen et al., 2014; Duquesne et al., 2020; Jamuar et al., 2014; Poirier et al., 2013; Cavallin et al., 2016; Michels et al., 2017). Neurodevelopmental disorder is the result of KIF5C coded kinesin proteins that host a non-functional motor domain and lose the ability to bind to hydrolyzed ATP (Padzik et al. 2016), which alters or inhibits the transport of cargo mediated by KIF5C along microtubules. It is interesting to note that five of the seven patients reported had variants involving Glu237 amino acid, four of which shared the same false-sense variant, p.Glu237Lys, suggesting a hot spot for variants and potential genotype-phenotype correlations (Michels et al. 2017).
To date, only three false-sense variants of KIF5C have been reported (Willemsen et al. 2014; Jamuar et al., 2014; Poirier et al., 2013; Cavallin et al., 2016; Michels et al., 2017). The KIF5C gene was first identified as the cause of cortical development malformation (Poirier et al. 2013). People affected generally have neurodevelopmental retardation, childhood epilepsy, intellectual impairment and psychomotor retardation and behavioural problems (Duquesne et al. 2020; Banerjee et al., 2024; Michels et al., 2017). Malformation of cortical development may include polymicrogyria and pachygyria and cerebral atrophy (Duquesne et al. 2020; Banerjee et al., 2024; Michels et al., 2017). However, clinical signs such as epilepsy, developmental delays and intellectual impairments are common clinical presentations for a wide variety of genetic and multifactorial conditions and therefore do not immediately suggest an underlying etiology of KIF5C. The increasing use of new generation sequencing technologies (NGS) in the diagnostic assessment of these children leads to the discovery of variants of uncertain significance in KIF5C. Here we report six other children with de novo variants in KIF5C, four of which have never been reported before. By adding these patients to the seven previously reported patients, the phenotypic spectrum associated with the pathogenic variants of KIF5C is expanded.
Method
This series of cases was prepared on the basis of protocol #19-0751 of the Colorado Multiple Institutional Review Board (COMIRB).
Results and conclusions
Four males from the same family with a disruptive mutation in the KIF4A bound to the X showed mild to moderate DI and epilepsy. A patient with novo false-sense mutation in KIF5C had severe ID, epilepsy, microcephaly, and cortical malformation. KIFA4 inactivation in primary rat hippocampus neurons altered the balance between excitatory and inhibitory synaptic transmission, while Kif5c mutation affected its protein function in excitatory synapses. Our results suggest that KIF4A and KIF5C mutations cause ID by tilting the balance between excitatory and inhibitory synaptic excitability.
__________
Link to the complete study : Wiley Online Library
Analysis on a panel of 30 patients
The following data are provided for information purposes by 30 families. In all cases, parents remain the experts of their child and it is shown that patients with KIF5C mutation all progress when stimulated and accompanied in appropriate therapies. These figures are therefore taken at a given moment, covering 30 children from 16 different countries (16 girls aged 2 to 27 and 14 boys aged 16 months to 19).
Age range of patients :
- Toddlers from 0 to 3 years 17%
- Children from 4 to 11 years 63%
- Teenagers aged 12-18 20%
- Adults 19-25 years 10%
- Adults over 25 years 7%
%
Non ambulatory
- Girls 23%
- Boys 77%
%
Non verbal
- Girls 52%
- Boys 48%
%
No solid food
- Girls 40%
- Boys 60%
%
Seizures
- Girls 50%
- Boys 50%
Types of care and support
How can I be accompanied ?
There is no specific treatment for this KIF5C mutation to date. Patient journeys are very different according to patient profile and needs, family resources, the country, but progress is possible when patients are stimulated and cared for. Families make a great job of accompanying the disease and often replace the missing care. Multidisciplinary support should therefore be provided as soon as possible.
Financial support provided by Health Security System in France :
Supported
- Neuropaediatric/pediatrician
- Endocrinologist
- Geneticist
- Ophthalmologist / orthoptist
- ENT
- Psychiatrist
- Physical Therapist
- Orthophonist
- Neurologist
- Doctor specializing in sleep disorders
- Doctor specializing in eating disorders
- Physician specializing in neurodevelopmental disorders (ASD, ADHD, PDD)
- Nurse / caregiver
Not supported
- Neuropsychologist
- Psychomotorian
- Occupational therapist
- Psychologist
- Specialist educator
- Nutritionist
- Osteopath
- Equitherapy/Hippotherapy
- Aquatherapy
HAPPY BENJI: Forward, walk! is a charity founded in 2025 by Benjamin's parents.
It works to gain recognition of the disease due to the gene KIF5C mutation , to unite families of patients affected and to convince the medical profession to undertake gene therapy research.
